Tools Available for Data Analysis and/or Visualization of 4DN-Related Datasets.
Here we compiled a list of tools available for data analysis and/or visualization of 4DN-related datasets.
If you have any suggestions to add/update this list, please send them to us via email: qiw024@ucsd.edu.
3D Genome Browser
With 3D Genome Browser you can join 50,000 other users from over 100 countries to explore chromatin interaction data, such as Hi-C, ChIA-PET, Capture Hi-C, PLAC-Seq, and more.

Genomic Interaction Visualization Engine
GIVE is an open source programming library that allows anyone with HTML programming experience to build custom genome browser websites or apps. With a few lines of codes, one can add to a personal webpage an interactive genome browser that host custom data. It typically takes less than half a day to build a genome browser website with GIVE.
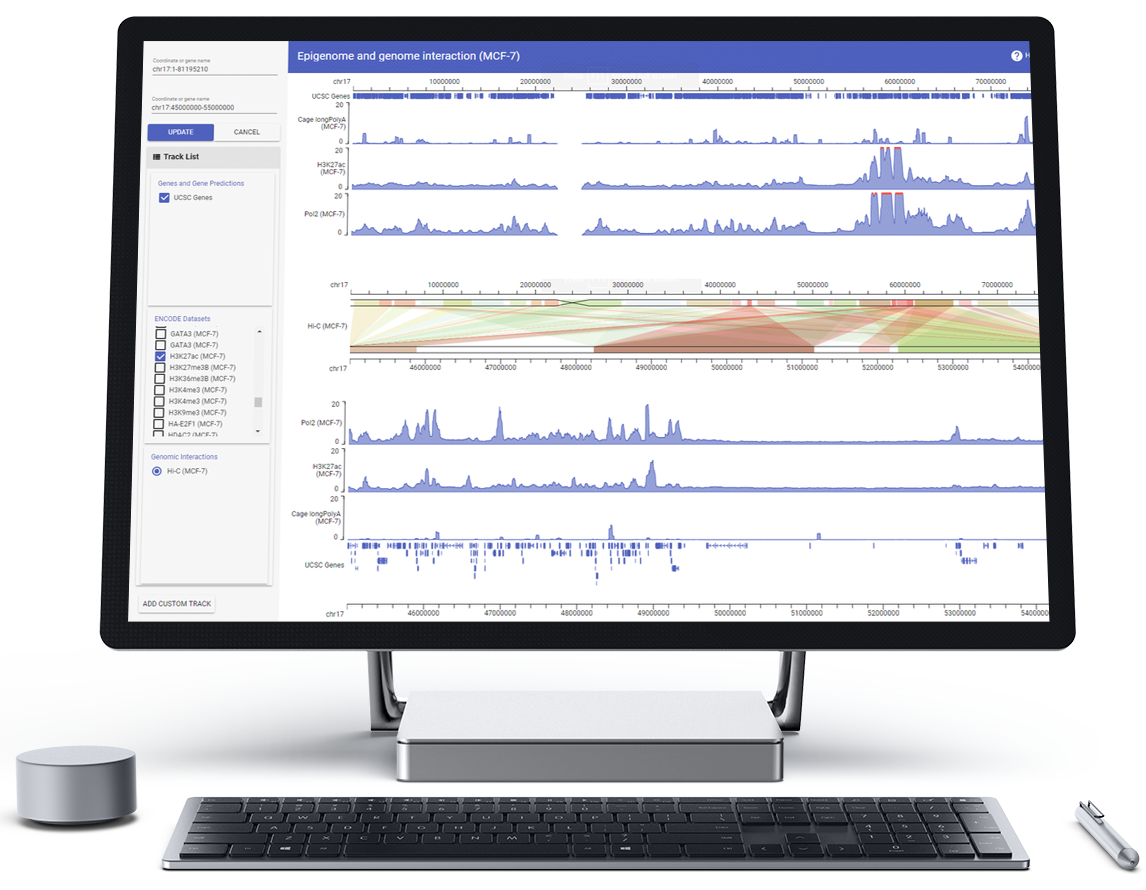
HiGlass
HiGlass is a web-based viewer for genome interaction maps featuring synchronized navigation of multiple views as well as continuous zooming and panning for navigation across genomic loci and resolutions. It provides a highly configurable user interface which facilitates the visual comparison of Hi-C and other genomic data from different experimental conditions. Examples and documentation can be found at HiGlass.
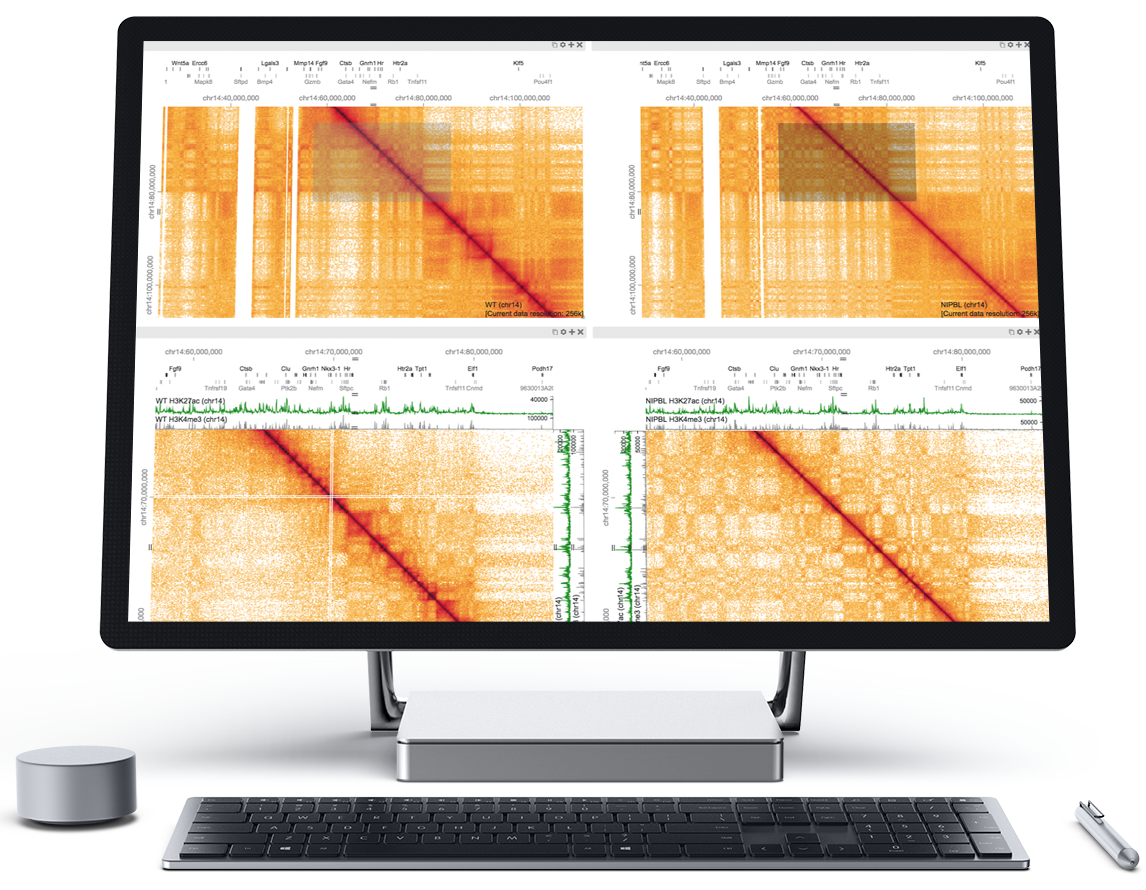
Juicer
Juicer is a one-click pipeline for processing terabase scale Hi-C datasets. Using Juicer, you can
- Go from raw fastq files to Hi-C maps binned at many resolutions
- Automatically annotate loops and contact domains with the Juicer tools
- Run the pipeline in the cloud, on LSF, Univa, or SLURM, or on a single CPU
Juicer creates hic files from raw (unaligned) reads derived from a Hi-C experiment.
Juicer Tools Pre creates hic files from aligned Hi-C reads (i.e., lists of Hi-C contacts).

Nucleome Browser
Nucleome Browser is a multimodal data exploration platform designed for systems biology study in this data science era. Nucleome Browser is a high-dimensional navigation map/system for exploring nucleome data, which includes genomics, 3D chromosome structure, imaging data and other future biotechnology data formats.

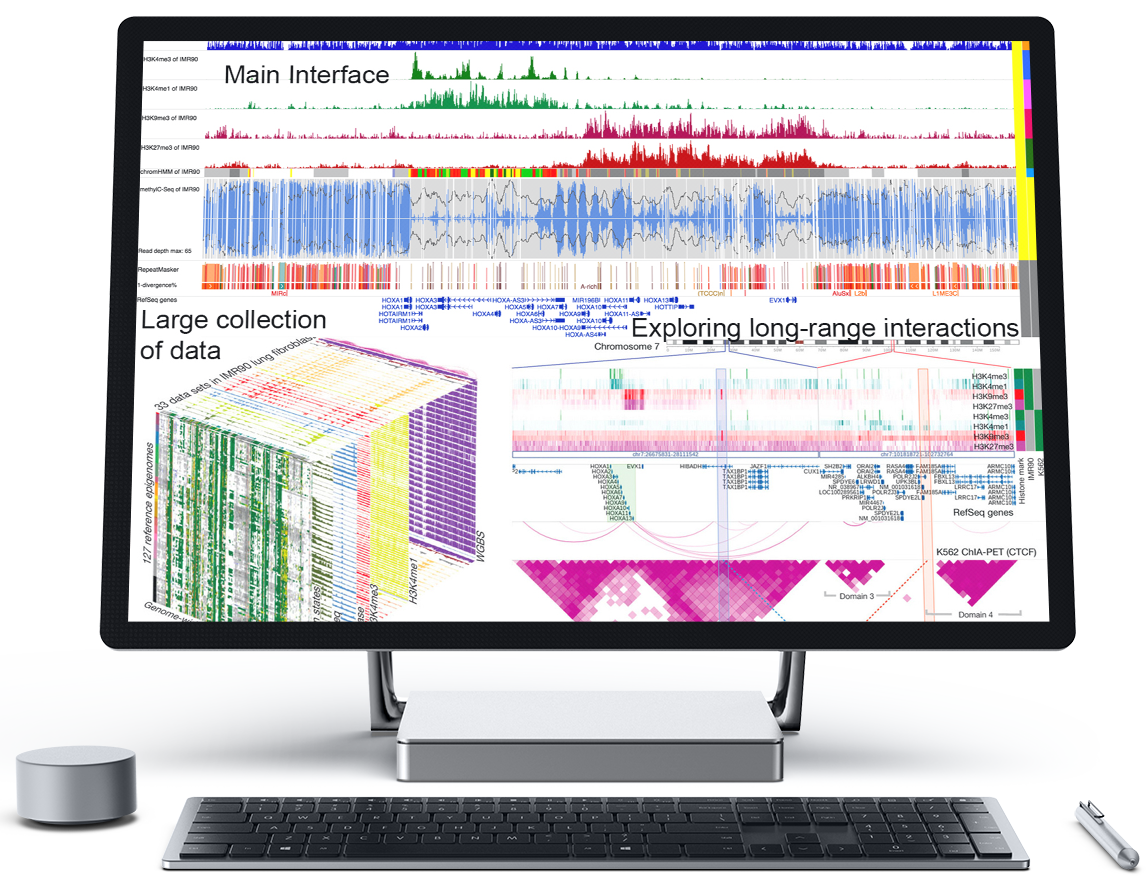
WashU Epigenome Browser
WashU Epigenome Browser is a web-based genomics browser which provides interactive visualization, integration, comparison and analysis of large genomics data. It allows investigators to explore epigenomic data in the context of higher-order chromatin interactions.
It hosts a large amount of datasets from international consortium including Roadmap, ENCODE, and 4DN projects, and supports user’s own data. The Browser can be easily installed locally or on Cloud services for investigating private data.
Software
| Software | Data Type | Description |
|---|---|---|
| Hi-C | A Computational Pipeline for Non-Random Interaction Identification, 3D Chromatin Structure Reconstruction from Hi-C Data and integrate it with eQTL Data. | |
Hi-C, Virtual 4C, ChIA-PET, Capture Hi-C, PLAC-seq, and more | With 3D Genome Browser you can join 50,000 other users from over 100 countries to explore chromatin interaction data, such as Hi-C, ChIA-PET, Capture Hi-C, PLAC-Seq, and more. | |
| 3DIV | Hi-C | 3DIV is a database which processed 315 Hi-C experiments from 80 human samples. 3DIV is an easy-to-use database providing epigenetic annotations and one-to-all chromatin interaction visualization with a variety of options. |
| 4C-ker | 4C | 4C-ker is a method developed to analyze 4C-Seq (circularized chromosome conformation capture) data. 4C-ker is a Hidden-Markov Model based pipeline that identifies regions throughout the genome that interact with the 4C bait locus. |
| AutoChrom3D | 3C, 4C, 5C, Hi-C | AutoChrom3D is an automatic pipeline to model chromatin regions with genomic size ranging from hundreds of kilobases to megabases at 8 kb resolution. |
| Centurion | Hi-C | A method that jointly calls all centromeres in a genome-wide Hi-C contact map. |
| ChIA-PET | A software package for automatic processing of ChIA-PET sequence data, including linker filtering, mapping tags to reference genomes, identifying protein binding sites and chromatin interactions, and displaying the results on a graphical genome browser. | |
| ChiaSig | ChIA-PET | ChiaSig finds significant interactions based on the NCHG model. |
| CHiCAGO | Hi-C | A set of tools for calling significant interactions in Capture Hi-C data, such as Promoter Capture Hi-C. |
| CHROMATIX | Hi-C | CHROMATIX is 3-D chromatin folding software for deconvolving population Hi-C into single-cell ensembles and identifying significant higher-order chromatin interactions. |
| ChromContact | Hi-C | ChromContact is a web server for analyzing spatial contact of chromosomes from the publicly available Hi-C data. |
| chromoR | Hi-C | chromoR provides users with a statistical pipeline for analysing chromosomal interactions data (Hi-C data). |
| ChromSDE | Hi-C | A deterministic method which applies semi-definite programming techniques to find the best structure fitting the observed data and uses golden section search to find the correct parameter for converting the contact frequency to spatial distance. |
| ChromStruct | Chromosome conformation capture | ChromStruct is a Python code to estimate the 3D chromatin structure from chromosome conformation capture data are presented. The only input required is a text file containing a general real matrix of contact frequencies. The method is based on a multiresolution, modified-bead-chain chromatin model, evolved through quaternion operators in a Monte Carlo sampling. |
| cooler | Hi-C | Cooler is a library for a scalable and interoperable storage format for genomic interaction data, built on HDF5. The cooler file type is a 4DN standard for binned genomic interaction data, e.g., Hi-C contact matrices. It supports flexible layouts for single-resolution (.cool), multi-resolution (.mcool), single-cell (.scool) datasets, and more. The library provides a paired command line interface (CLI) and Python application programming interface (API) for efficient creation, querying, manipulation, and normalization of contact matrices. |
| cooltools | Hi-C | Cooltools leverages the widely-adopted cooler format to enable analysis of high-resolution Hi-C datasets. Cooltools provides a paired command line interface (CLI) and Python application programming interface (API), which respectively facilitate workflows on high-performance computing clusters and in interactive analysis environments. |
| CytoHiC | Hi-C | A Cytoscape plugin, which allow users to view and compare spatial maps of genomic landmarks, based on normalized Hi-C datasets. |
| diffHiC | Hi-C | A software package for the detection of differential interactions from Hi-C data. diffHic provides methods for read pair alignment and processing, counting into bin pairs, filtering out low-abundance events and normalization of trended or CNV-driven biases. |
| distiller-nf | Hi-C | Distiller is a Nextflow pipeline specialized for scalable processing of 3C-derived sequencing data, e.g., Hi-C or Micro-C. Distiller performs read alignment, classifies sequencing alignments into contact pairs using pairtools, filters amplification duplicates, and aggregates pairs into binned and normalized contact matrices with cooler. |
| Edison | Genome sequences (FASTA) or genome assemblies (AGP) | Edison uses a graph based edit distance calculation to find the lowest number of edits needed to reorganize one genome into another. It was developed in the context of measuring scaffolding accuracy, and given a reference genome, it can determine how well scaffolds are able to organize contigs into their appropriate locations in chromosomes. |
| EpiAlignment | Bigwig, Genome annotations | EpiAlignment is a dynamic programming algorithm for chromosomal similarity search, which is able to align two regions with both sequences and epigenomic information, such as histone modifications. |
| FastHiC | Hi-C | FastHiC is a fast peak caller for Hi-C data. |
| FisHiCal | Hi-C | FisHiCal integrates Hi-C and FISH data, offering a modular and easy-to-use tool for chromosomal spatial analysis. |
| Fit-Hi-C | Hi-C | Fit-Hi-C is a tool for assigning statistical confidence estimates to intra-chromosomal contact maps produced by genome-wide genome architecture assays such as Hi-C. |
| G-Dash | Bigwig, 2Bit, 3D models in xyz/pdb | G-Dash is a genome dashboard that integrates the informatics capabilities of a genome browser with controllers for generating and navigating 3D models of DNA, nucleosomes and chromatin from base pairs to mega base pairs and beyond. |
| Hi-C | A Galaxy based web server to give biomedical researchers an easy to use access to the HiCExplorer (for more information see the related item below in the table) and additional high-throughput analysis software like deeptools or mappers like BWA-MEM or Bowtie are provided. | |
| Hi-C, ChIA-PET | R package for handling Genomic interaction data, such as ChIA-PET/Hi-C, annotating genomic features with interaction information and producing various plots/statistics. | |
| GITAR | Hi-C | GITAR (Genome Interaction Tools and Resources) is a standardized way to analyze and visualize genomic interaction data. The tool is composed of two modules: HiCtool (a standardized Python library for processing and visualizing Hi-C data) and processed data (a collection of datasets processed using HiCtool). HiCtool performs also A/B compartment and TAD analysis. |
| GIVE | Hi-C, ChIA-PET | GIVE is an open source programming library that allows anyone with HTML programming experience to build custom genome browser websites or apps. With a few lines of codes, one can add to a personal webpage an interactive genome browser that host custom data. It typically takes less than half a day to build a genome browser website with GIVE. |
| GMAP | Hi-C | GMAP (Gaussian Mixture model And Proportion test), is an algorithm to identify topologically associating domains and subdomains (Yu et al., Nature Communications, 2017). It provides a new and efficient tool to analyze simulated and experimental Hi-C data with increased statistical robustness and better consistency of the predictions compared to three state-of-the-art methods |
| GOTHiC | Hi-C | A Hi-C analysis package using a cumulative binomial test to detect interactions between distal genomic loci that have significantly more reads than expected by chance in Hi-C experiments. |
| Hi-C | The tool enables the generation of genome-wide 3D proximity maps. You will also be able to visually navigate the dataset and explore Hi-C proximity maps of entire human genomes. | |
| Hi-C Pipeline | Hi-C | Hicpipe is a set of scripts and programs that correct Hi-C contact maps, given a list restriction enzyme sites and mapped paired reads. |
| Hi-Corrector | Hi-C | A fast, scalable and memory-efficient package for normalizing large-scale Hi-C data. |
| HiBrowse | Hi-C | A statistical web toolkit for analyzing, interpreting and visualizing genome-wide chromosome conformation capture data, such as Hi-C, TCC, GCC and similar. |
| HiC-ACT | Hi-C | HiC-ACT is a Hi-C peak caller via aggregated Cauchy test. |
| HiC-bench | Hi-C | HiC-bench is a tool for a comprehensive and reproducible Hi-C data analysis designed for parameter exploration and benchmarking. |
| HiC-inspector | Hi-C | A bioinformatics pipeline for the automated analysis of data generated by high-throughput chromatin conformation capture (HiC). |
| HiC-Pro | Hi-C | HiC-Pro is an optimized and flexible pipeline for processing Hi-C data from raw reads to normalized contact maps. |
| HiCapp | Hi-C | HiCapp is a Hi-C analysis pipeline which can correct for the copy number bias in tumor Hi-C data using caICB correction algorithm. |
| HiCAR | HiChIP, ChIA-PET, PLAC-seq | HiCAR (HiC on Accessible Regulatory DNA) is a robust and sensitive assay for measurement of chromatin accessibility and cis-regulatory chromatin contacts. Different from immunoprecipitation-based methods, HiCAR does not require antibodies. |
| HiCdat | Hi-C | HiCdat provides a simple graphical user interface for data pre-processing and a collection of higher-level data analysis tools implemented in R. |
| HiCExplorer | Hi-C | Hi-C analysis software which covers the whole pipeline of Hi-C data analysis. Starting with the creation of the contact matrix, the correction, computation of TADs and A / B compartments to a visualization of these. Additional data tracks from other high-throughput analysis like ChIP-Seq or gene tracks can be added to the visualization. |
| HiCNorm | Hi-C | A parametric model to remove systematic biases in the raw Hi-C contact maps, resulting in a simple, fast, yet accurate normalization procedure. |
| HiCPlotter | Hi-C | An easy-to-use open-source visualization tool to facilitate juxtaposition of Hi-C matrices with diverse genomic assay outputs, as well as to compare interaction matrices between various conditions. |
| HiCPlus | Hi-C | HiCPlus uses deep learning to enhance Hi-C data resolution. |
| HiCRep.py | Hi-C | HiCRep.py is an efficient python implementation of the HiCRep framework for evaluating the reproducibility of Hi-C matrices. |
| HiCRes | Hi-C | HiCRes predicts the resolution (as defined in the Rao et al paper from 2014) that small HiC libraries will reach at deeper sequencing level. It works by modeling the read distribution on the chromatin and it is extremely accurate. |
| HiCseg | Hi-C | This package allows you to detect domains in HiC data by rephrasing this problem as a two-dimensional segmentation issue. |
| hiCtools | Hi-C | HiC is high-throughput sequencing technology applied to chromatin conformation techniques. This collection of tools stream-lines the processing of HiC data from raw sequence to contact matrices and beyond. |
| HiCUP | Hi-C | A tool for mapping and performing quality control on Hi-C data. |
| HiFive | 5C, Hi-C | The HiFive tool suite provides efficient data handling and a variety of normalization approaches for easy, fast analysis and method comparison. |
| HiGlass | Interaction matrices | HiGlass is a tool for displaying and comparing large matrices within a web page. Matrices are typically visualized as heatmaps. In the case of large matrices, the heatmaps are too large to render all at once. Instead, we aggregate their values and display summaries at lower resolution while allowing one to zoom in and explore them in greater detail. |
| HiPiler | Hi-C | HiPiler is an interactive web application for feature-centric exploration of Hi-C matrices. HiPiler represents 2D features or annotations (like loops, TADs, CTCF sites, etc.) as individual thumbnail-like snippets. Snippets can be laid out automatically based on their data and meta attributes. They can be interactively aggregated into piles for stratification and are visually linked back to the matrix to provide context. |
| HIPPIE | Hi-C | HIPPIE is the workflow for analyzing batches of Hi-C paired-end reads in compressed FASTQ format (.fastq.gz) and predict enhancer-target gene interactions. |
| HiTC | 5C, Hi-C | The package explores high-throughput 'C' data such as 5C or Hi-C. |
| HMRF | Hi-C | A hidden Markov random field based Bayesian peak caller to identify long range chromatin interactions from Hi-C data. |
| HOMER | Hi-C | HOMER contains several programs and analysis routines to facilitate the analysis of Hi-C data. |
| HPRep | PLAC-seq, HiChIP | HPRep is a tool for evaluating reproducibility of PLAC-seq and HiChIP data. |
| HubPredictor | Hi-C | A computational model integrating Hi-C and histone mark ChIP-seq data to predict two important features of chromatin organization: chromatin interaction hubs and topologically associated domain (TAD) boundaries. |
| HUGIn | Hi-C | HUGIn is an integrative Hi-C data visualization tool. |
| ICE | Hi-C | A computational pipeline that integrates a strategy to map sequencing reads with a data-driven method for iterative correction of biases, yielding genome-wide maps of relative contact probabilities. |
| Juicebox | Hi-C | Software for visualizing data from Hi-C and other proximity mapping experiments. |
| Juicer | Hi-C | Juicer is a one-click system for analyzing loop-resolution Hi-C experiments. Specifically, the software is an end-to-end automated pipeline for converting raw reads into Hi-C maps and loop calls using only a single command. |
| LACHESIS | Hi-C | A software tool to measure the thread of life. LACHESIS exploits contact probability map data (e.g. from Hi-C) for chromosome-scale de novo genome assembly. |
| LSMMD-MA | scRNA; scATAC; scHiC; visual cell sorting imaging and sequencing (sciRNA; sciATAC; sciHiC) | LSMMD-MA is a large-scale Python implementation of the MMD-MA method for multimodal data integration. By reformulating the MMD-MA optimization problem using linear algebra and solving it with KeOps, a CUDA framework for symbolic matrix computation in Python, LSMMD-MA scales to a million cells in each modality, two orders of magnitude greater than existing implementations. |
| Mango | ChIA-PET | A complete ChIA-PET data analysis pipeline that provides statistical confidence estimates for interactions and corrects for major sources of bias including differential peak enrichment and genomic proximity. |
| MAPS | PLAC-seq, HiChIP | MAPS is a pipeline to identify significant long-range chromatin interactions from PLAC-seq and HiChIP datasets. |
| MDM | ChIA-PET | MDM provides functions for the analysis of chromatin interactions using MC_DIST model, Two-Step model and One-Step model, based on ChIA-PET data. |
| MIA-Sig | ChIA-Drop | MIA-Sig is a python package for calling statistically significant multiplex chromatin complexes from non-enriched ChIA-Drop and protein-enriched ChIA-Drop datasets. |
| MICC | ChIA-PET | An easy-to-use R package to detect chromatin interactions from ChIA-PET sequencing data. By applying a Bayesian mixture model to systematically remove random ligation and random collision noise, MICC could identify chromatin interactions with a significantly higher sensitivity than existing methods at the same false discovery rate. |
| MOGEN | Hi-C | A 3D chromosome reconstruction method to make it capable of reconstructing 3D models of genomes from both intra- and inter-chromosomal Hi-C contact data. |
| NADfinder | NAD-seq | NADfinder is a Bioconductor package for bioinformatic analysis of the NAD-seq data, including normalization, smoothing, peak calling, peak trimming and annotation. |
| NuChart | Hi-C | An R Package to Study Gene Spatial Neighbourhoods with Multi-Omics Annotations. |
| Multi-data | Nucleome Browser is a multimodal, interactive data visualization and exploration platform to integrate genomic, imaging, and 3D structure data. | |
| pairtools | Hi-C | Pairtools offers a comprehensive and modular suite for identifying and manipulating contact pairs from proximity ligation sequencing experiments. Pairtools takes in sequencing alignments and outputs 4DN standard .pairs files. Pairtools offers additional filtering, manipulation, and quality control within the same framework. |
| PartSeg | 3DFish | PartSeg allows super-resolution and 3DFish imaging data segmentation and analysis in the batch mode for hundreds/thousands of cells. This application is designed to help biologist with segmentation based on threshold and connected components. |
| PASTIS | Hi-C | PASTIS is a software tool for inferring a consensus model of 3D genome architecture from Hi-C data. It can model multiple chromosomes at once and can model haploid or diploid genomes. |
| Polarbear | scRNA-seq and scATAC-seq (co-assay and single-assay) | Polarbear is a semi-supervised deep neural network method for single cell cross-modality translation. |
| R3CPET | ChIA-PET | The package provides a method to infer the set of proteins that are more probably to work together to maintain chormatin interaction given a ChIA-PET experiment results. |
| SnapHiC | Hi-C | SnapHiC: Single nucleus analysis pipeline for Hi-C data is a tool to identifying chromatin loops from single cell Hi-C data. |
| Synmatch | single-cell multi-omics data: scRNA, scATAC, scHi-C, scMethyl, etc. | Synmatch is a software tool for finding a direct matching of cells from different single-cell measurements. It takes as input two matrices of single-cell profiles measuring different cellular properties, such as gene expression and chromatin accessibility, and outputs a matching of the cells across the datasets. The key idea behind Synmatch is that the same cell, when measured in two different modalities, is likely to have similar sets of neighboring cells in the two spaces. This intuition is used to formulate the matching problem as a supermodular optimization over the neighborhood structure of the two modalities, and the problem is solved by a fast greedy heuristic which scales to thousands of cells. The two modalities need not share any features, Synmatch operates in an entirely unsupervised manner. |
| TADbit | Hi-C | An algorithm for the identification of hierarchical topological domains in Hi-C data. |
| TADsplimer | Hi-C | TADsplimer is a computational tool to systematically detect topologically associating domain (TAD) splits and mergers across the genome between Hi-C samples. |
| TADtree | Hi-C | TADbit is a complete Python library to deal with all steps to analyze, model and explore 3C-based data. |
| THUNDER | Hi-C | THUNDER is a deconvolution method for bulk Hi-C data. |
5C, Hi-C, ChIA-PET | The browser supports multiple types of long-range genome interaction data. This enables investigators to explore epigenomic data in the context of higher-order chromosomal domains and to generate multiple types of intuitive, publication-quality figures of interactions. |